编者按:日前,EHA 2025科学会议报告正式发布,涵盖恶性血液学与非恶性血液学两大部分!报告全面回顾了在EHA 2025大会上展示的最重要科学进展与临床见解,为全球血液学专业人士提供了深入了解血液学各大领域最新动态的机会。本期特整理恶性血液学报告中关于髓系肿瘤的精粹内容,旨在为广大同行提供有价值的临床与科研洞见。
精选演讲概览

开发模型以更好地理解MDS的发病机制和克隆演化
骨髓增生异常综合症(MDS)是一种与异常血细胞生成相关的克隆性血液疾病。其特征包括外周血细胞减少、凋亡增加以及骨髓原始细胞数量增加。MDS的5年生存率约为30%,且发病率随年龄增长而增加[1]。潜能未明的克隆性造血 (CHIP)是一种常见的年龄相关现象,可进展为MDS。多种突变可以导致MDS,包括与SF3B1突变相关的MDS环形铁粒幼红细胞(MDS-RS)亚群。随着亚克隆中其他突变的获得,最终可能发展为急性髓性白血病(AML)[2]。
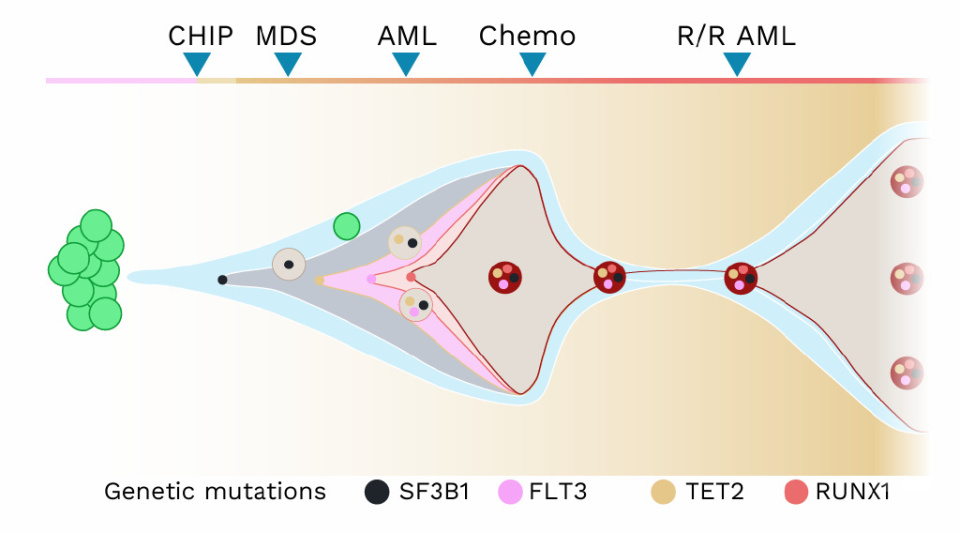
进行性髓系恶性肿瘤中的克隆选择
为了能够早期干预并通过治疗手段阻止疾病的演化,研究人员需要可靠的小鼠模型来研究MDS。使用携带MDS突变的人类间充质干细胞(MSC)在免疫缺陷小鼠中进行研究的成功率较低。然而,Bonnet教授的团队开发了一种方法,通过使用基于明胶的支架与MSC和内皮祖细胞结合,这种方法报告称MDS骨髓CD45+移植的成功率为1%到40%,在某些情况下甚至高达80%[3]。这一模型有助于识别维持MDS细胞存活的因素,预测患者对治疗的反应,并研究疾病随时间变化的过程,以改善MDS的治疗选择。
探索DDX41致病突变携带者在MDS/AML(MN)中的风险
了解MDS等髓系肿瘤(MNs)的发病机制和克隆演化非常重要,同时也必须了解患者的易感性和患病风险,以便能够识别患者并量身定制治疗。例如,估计5%至10%的髓系肿瘤患者存在遗传性基因易感性[4]。其中DDX41基因的胚系突变最为常见,约占MDS/AML病例的5%。携带DDX41胚系突变的患者通常在发病年龄上与散发性病例相似,但与患有野生型DDX41的MDS或AML患者相比,其生存期更长[5]。
在选择造血干细胞移植供体(通常是亲属)时,需要考虑该MN亚群的遗传性。越来越多的证据表明,如果供者携带DDX41致病突变,那么复发的风险会增加。根据一项回顾性日本亲属队列研究,40岁之前的绝对风险较低,但在90岁时风险可升高至49%。此外,研究还指出,携带DDX41截断突变的MDS患者进展为AML的速度较快[6]。相比之下,英国生物库研究显示,普通人群中MDS/AML的绝对风险仅为男性5.5%,女性1.37%[7]。这些发现为携带者的监测建议提供了依据。为此,Villy等[8]开展了覆盖法国11个中心的项目,旨在识别并监测携带DDX41胚系突变的家庭,评估携带者发生MDS/AML的累积风险。相关结果目前正在《European Journal of Human Genetics》审稿中。对于更大样本量和更具代表性人群的进一步研究仍是必要的。
MPN中的克隆选择及其后果
与其他髓系肿瘤一样,骨髓增殖性肿瘤(MPN)也可以通过克隆演化进展为继发性AML。这一过程由Janus激酶(JAK)信号转导激活突变和克隆适应性驱动[9-10]。许多突变已被描述为预测总生存率的预后因素(如K/NRAS[11]、NFE2[12]和TP53[13-14]),以及动脉血栓形成(如TET2或DNMT3A)和治疗耐药性(如TET2、DNMT3A、ASXL1、EZH2、IDH1/2);然而,年龄是目前已知的唯一进展风险因素。某些克隆的扩增并非仅仅由新突变的获得,微环境也在其中发挥着重要作用,且有时还会受到治疗的影响。例如,JAK抑制剂芦可替尼(ruxolitinib)通过释放RAS突变克隆,使其免于致癌基因诱导的衰老,来激活这些克隆。新的数据表明[15],当比较未接受芦可替尼治疗的无RAS通路突变患者与接受芦可替尼治疗的RAS通路突变患者时,无转化生存期无显著差异(P=0.555)。然而,接受芦可替尼治疗的RAS通路突变患者的无转化生存期显著短于接受芦可替尼治疗的无RAS通路突变患者(P<0.0001)。这表明治疗暴露可以驱动MPN中的克隆演化,而对克隆结构的评估可能有助于改善预后评估并指导未来的治疗选择。
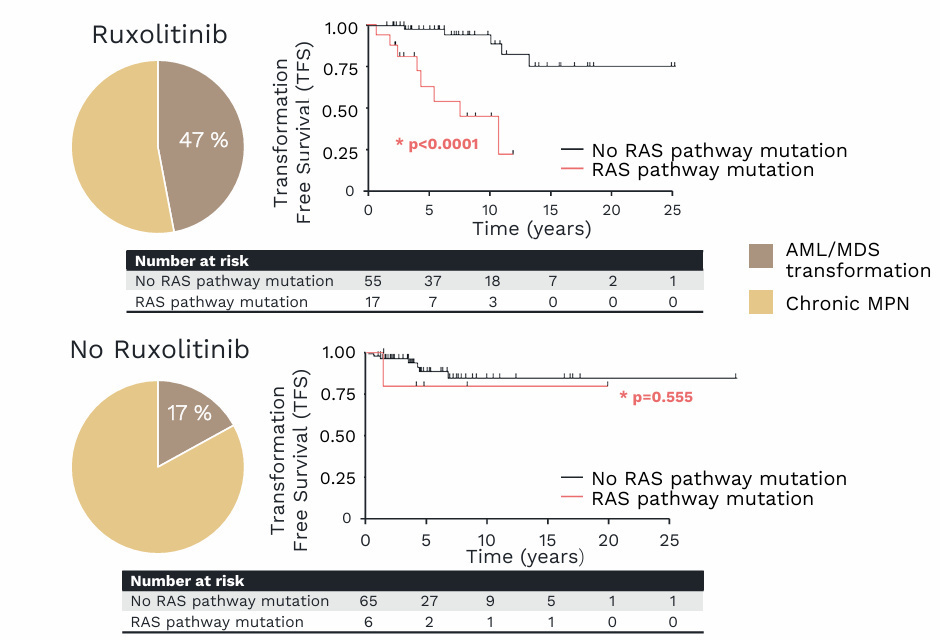
芦可替尼(Ruxolitinib)促进了RAS突变克隆的扩增,并加剧了RAS突变带来的风险
抗体靶向突变CALR在MPN中的应用
突变钙网蛋白(mutCALR)是MPN中的第二大驱动突变[16-17],可产生一种新的C末端尾部,从而激活质膜上的MPL/JAK2/STAT信号通路,并促进癌性增殖。由于内质网滞留序列 (KDEL) 的存在,野生型 CALR 蛋白无法定位在细胞表面,而该序列在 CALR-mut 中丢失。细胞表面CALR是MPN的标志物,可以选择性地靶向。另一方面,大约四分之一的原发性血小板增多症(ET)患者——一种MPN——携带CALR突变,并且大多数患者的突变属于两种类型(Type 1和Type 2)[18-20]。 这些患者通常有较高的转化为骨髓纤维化的风险,但目前的治疗并未靶向这些驱动突变[21]。当前有两种主要策略正在探索中,并在EHA 2025大会上展示了相关进展[22]。
首先,靶向致癌性mutCALR/MPL的治疗策略,包括使用INCA33989(一种单克隆抗体)来抑制STAT信号通路,防止癌性增殖并诱导细胞凋亡,从而使巨核细胞生成正常化,减少致病细胞,降低血小板增多症并防止白血病的出现。INCA33989目前正在进行临床评估。在两项1期剂量递增研究(NCT05936359——美国外,NCT06034002——仅限美国)中,共有49名ET患者接受了INCA33989的治疗,剂量范围从24 mg到2500 mg。未发现剂量限制性毒性,但有3例发生了≥3级的脂肪酶水平升高,且被归类为严重不良事件。大多数患者的血小板计数在接受治疗后迅速并持续地恢复正常,特别是在剂量≥400 mg时。携带Type 1突变的患者在较低剂量下有较好的反应,而在较高剂量(≥400 mg)下,携带Type 2突变的患者也表现出较好的疗效。生物标志物分析显示,突变的干/祖细胞和巨核细胞数量减少。Mascarenhas等[23]总结道:“这些发现支持INCA33989在为突变CALR引起的ET患者提供持久血液学反应和改善疾病的潜力。”
其次,针对突变CALR的T细胞招募也在EHA 2025大会上进行了展示,使用JNJ-88549968和INCA035784(mutCALR×CD3抗体)来诱导选择性细胞毒性[22,24]。临床前研究和小鼠体内模型表明,JNJ-88549968与肿瘤体积缩小和生存获益相关,促使了1期临床试验(NCT06150157)的启动[22]。通过使用TF-1母细胞系开发的细胞系,测试了INCA035784对不同形式mutCALR的选择性[10]。添加健康供体的T细胞,可以测试T细胞的激活、T细胞介导的毒性及其增殖。在研究中,INCA035784选择性地结合Type 1和Type 2突变CALR表达的工程TF-1克隆并激活了T细胞介导的功能。它不与野生型CALR的表面暴露部分结合,也不诱导健康供体外周血单核细胞中与细胞因子释放综合征相关的非特异性细胞因子分泌。在异种移植模型中治疗后,骨髓中的髓系细胞(CD33+)和巨核细胞(CD45+CD41+)减少。Psaila等[24]总结道:“总体而言,对于缺乏治愈性治疗选择的MPN患者,INCA035784代表了一种有前景的治疗方法。”
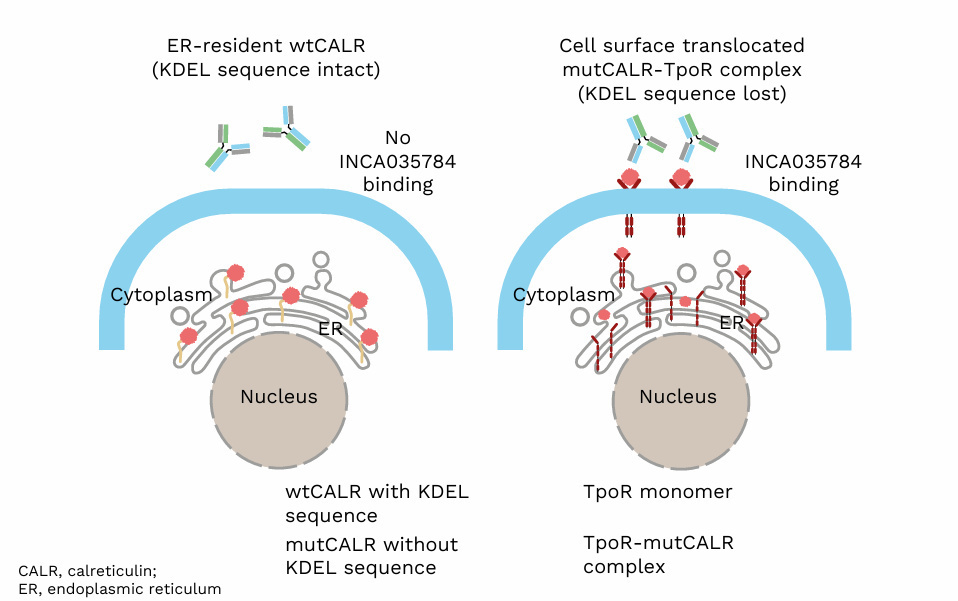
突变CALR在KDEL序列(内质网滞留信号)丧失后转移至细胞表面
新型抗体药物偶联物(ADC)选择性地将SMARCA2/4降解剂输送至MPN
mutCALR不仅因其在信号转导中的作用而成为一个有价值的靶点,还可以用于通过抗体药物偶联物(ADC)选择性地靶向细胞。失调的SWI/SNF活性已被与AML、MDS和MPN发生机制相关,SWI/SNF ATP酶SMARCA2和SMARCA4是MPN中的关键治疗靶点。研究人员发现了一种非拮抗、内化的CALR抗体,并以此为基础开发出一种新型ADC,这种ADC可以选择性地在CALR突变细胞中内化,而不在健康的野生型细胞中内化。CALR精准ADC(pADC)表现出选择性降解SMARCA2/4并引发CALR突变细胞的细胞毒性。可溶性突变CALR蛋白的存在不会影响CALR pADC的细胞毒性,因此似乎不会对疗效或安全性构成风险。测试的CALR pADC在体内(小鼠模型)展示了强大的抗肿瘤活性,并且耐受性良好,能够选择性地靶向并消除突变的外周疾病细胞,同时保护健康细胞。类似的结果也在CDK9降解CALR pADC中观察到,表明这种模式在多种载体中具有广泛的潜力[25]。
SANRECO:一项正在研究Divesiran治疗PV的1期临床试验
真性红细胞增多症(Polycythemia Vera,PV)表现为红细胞过度生成、铁缺乏和通常低血铁素水平[26]。 Divesiran是一种靶向TMPRSS6的新型GalNAc共轭siRNA,旨在提高PV患者血铁素水平,促进铁的重新分布,并限制其用于红细胞生成。由Kreyanskaya等[27]在EHA 2025大会上报告的1期试验结果(N=21)表明,divesiran治疗可降低治疗和随访期间的静脉放血率。所有剂量组的患者均观察到血红蛋白和红细胞比容的降低,且血铁素和铁蛋白水平呈剂量依赖性增加。治疗耐受性良好,没有出现剂量限制性毒性。最常见的治疗相关不良事件为注射部位反应、贫血和疲劳。正在进行的随机、双盲、Ⅰ/Ⅱ期研究(NCT05499013)目前正在评估患者在没有放血的情况下,血红蛋白比容是否能达到≤45%,同时改善与PV相关的症状[27]。
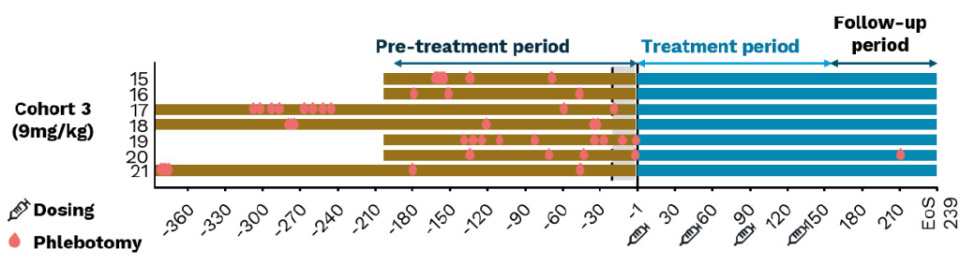
在治疗前和治疗期间,第3组(9 mg/kg剂量)中的放血率
在EHA2025大会上呈现的最新数据表明,对MPN分子机制的理解不仅持续深入,而且有助于开发更具靶向性的在早期阶段治疗恶性肿瘤的治疗方案。
参考文献:
1.Zeidan AM, et al. Epidemiology of myelodysplastic syndromes: Why characterizing the beast is a prerequisite to taming it. Blood. 2019;34:1-15.
2.Mian SA, et al Spliceosome mutations exhibit specific associations with epigenetic modifiers and proto-oncogenes mutated in myelodysplastic syndrome. Haematologica. 2013;98(7):1058-1066.
3.Bonnet D. Pathophysiology and clonal evolution in MDS. Oral presentation p115-1 at EHA2025.
4.Khoury JD, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36(7):1703-1719.
5.Sébert M et al. Germline DDX41 mutations define a significant entity within adult MDS/AML patients. Blood. 2019;134(17):1441-1444.
6.Makishima H, et al. Germ line DDX41 mutations define a unique subtype of myeloid neoplasms. Blood. 2023;141(5):534-549.
7.Cheloor Kovilakam S, et al. Prevalence and significance of DDX41 gene variants in the general population. Blood. 2023;142(14):1185 1192.
8.Villy MC. Myeloid neoplasms risks for germline DDX41 pathogenic variants carriers. Oral presentation S145 at EHA2025.
9.Jaiswal S. et al. Age-related Clonal Hematopoiesis Associated with Adverse Outcomes. N Engl J Med. 2014;371:2488-2498.
10.Genovese G. et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N Engl J Med. 2014;371:2477-2487.
11.Santos FPS, et al. Prognostic impact of RAS-pathway mutations in patients with myelofibrosis. Leukemia. 2020;34(3):799-810.
12.Marcault C et al. Impact of NFE2 mutations on AML transformation and overall survival in patients with myeloproliferative neoplasms. Blood. 2021;138(21):2142-2148.
13.Gagelmann N et al. Impact of TP53 on outcome of patients with myelofibrosis undergoing hematopoietic stem cell transplantation. Blood. 2023;141(23):2901-2911.
14. Rolles B et al. TP53 mutations and Their Impact on Survival in Patients with Myeloproliferative Neoplasms. ASH 2023:3160.
15. Benajiba L. Clonal selection in MPN and its consequence. Oral presentation p266-1 at EHA2025.
16. Klampfl T, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379-2390.
17. Nangalia J, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391-2405.
18. Guglielmeli P et al. CALR mutation burden in essential thrombocythemia and disease outcome. Blood. 2024;143:1310-1314.
19. Klampfl T et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369:2379-2390.
20. Nangalia J et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369:2391-2405.
21. Yacoub et al. Disease and Clinical Characteristics of Patients With a Clinical Diagnosis of Essential Thrombocythemia Enrolled in the MOST Study. Clin Lymphoma Myeloma Leuk. 2021;21:461-469.
22. Plo I. Antibody targeting of mutant CALR in MPN. Oral presentation p114 2 at EHA2025.
23. Mascarenhas J. INCA33989 is a novel, first in class, mutant calreticulin-specific monoclonal antibody that demonstrates safety and efficacy in patients with essential thrombocythemia (ET). Oral presentation LBA4002 at EHA2025. 24. Psaila B. INCA035784, a novel, equipotent T cell–redirecting antibody for patients with myeloproliferative neoplasms carrying different types of calreticulin mutations. Oral presentation S212 at EHA2025.
25. Fultang N. Discovery of first-in-class precision antibody drug conjugates targeting mutant calreticulin for the treatment of myeloproliferative neoplasms. Oral presentation S211 at EHA2025.
26. Ginzburg YZ, et al. Dysregulated iron metabolism in polycythemia vera: etiology and consequences. Leukemia. 2018;32:2015-2116.
27. Kremyanskaya M. SANRECO, an on going Phase1/2 study evaluating divesiran, a novel GalNAc-conjugated siRNA, in patients with polycythemia vera. Oral presentation S224 at EHA2025.